If you have landed on this page it is highly likely that you’re an A-Level student, or someone of a similar level, and you’re interested in taking your understanding of the neurological disorder ‘Amnesia’ to the next level.
If you feel that your current understanding of amnesia isn’t of an A-Level standard, or you feel you might need a quick refresher on the fundamental understanding of amnesia (particularly retrograde and anterograde), then the below link may help you get up to speed before continuing this article.
https://gcsepsychology.com/brain-damage-retrograde-anterograde-amnesia/
To better help apply the information in this wiki to A-Level studies, two different amnesia disorders will be referred to for examples on each of the key pages. These are: Wernicke-Korsakoff Syndrome (WKS) and Transient Global Amnesia (TGA). Other disorders may be referenced throughout but the three mentioned above will be the focus.
To fully understand amnesia, the processing of memories must first be understood. Briefly, short-term memory (STM) is defined as information held within conscious awareness, and which is currently receiving attention; it is of course contrasted to long-term memory (LTM) which is held in permanent storage available for retrieval at some time in the future (Groome, 2013, p. 137). For example, when revising for an exam the information the student is currently studying would be present in their short-term memory but the information they learnt in class at the start of the year that they haven’t revised yet would likely be in their long-term memory stores, if not forgotten.
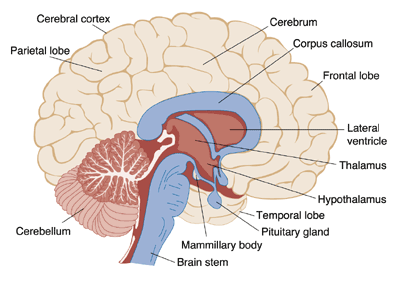
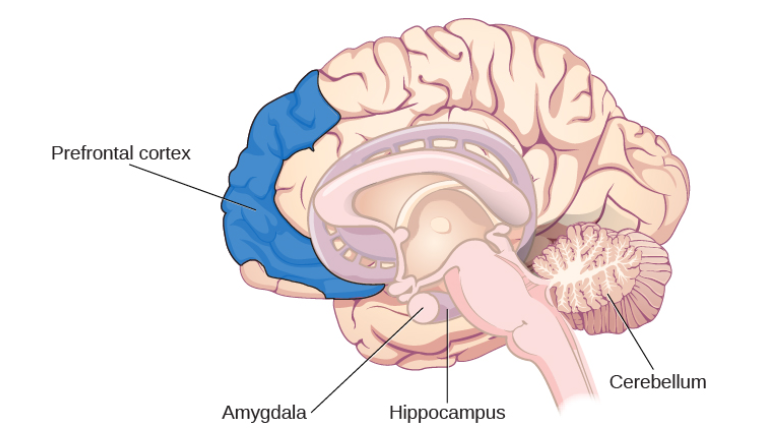
Many researchers believe that the entire brain is involved with memory, in some way or another (Lashley, 1950). However, some key areas have been identified (shown below in Figure 1.) and include the amygdala, hippocampus, cerebellum, the prefrontal cortex and the synapses linking them all (Mayford, Siegelbaum, & Kandel, 2012).
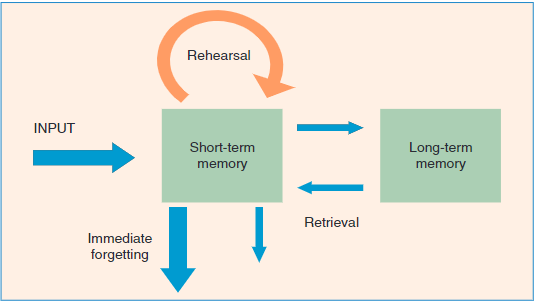
Forming memories involves various steps and stages (Atkinson & Shiffrin, 1968). To begin with the brain receives sensory information from several sources in different formats, such as smell, taste, sound, vision and touch. At this point the information that has the individuals focus will be rehearsed in the STM store and ‘encoded’ (transformed into a format the brain can retain). Following this, the information is then ‘committed to memory’ and stored in the LTM, readily available to be retrieved when needed. The information not rehearsed or not regularly retrieved will eventually be forgotten. Figure 2. Below illustrates this process.
Amnesia is essentially the instances where memory, or the process of forming memories, fails. The processes of forming, maintaining and recalling memories is extremely complex and as mentioned above involves numerous brain structures, if not the brain in its entirety. This can lead to numerous points of failure and thus numerous reasons for memory to fail; leading to amnesia.
In terms of understanding amnesia and memory, the convoluted and complex nature of the memory process can make it difficult to pinpoint which areas are responsible for which functions, this problem is also relevant when looking at the symptoms of amnesia which quite often include several deficits outside those of memory. These are referred to as ‘comorbid’ symptoms, diseases or conditions that occur alongside another disorder, and can make it difficult to differentiate the issues that are a direct result of amnesia and those that are not. Examples of those that appear alongside amnesia would be perceptual deficits and confabulation; this is often associated with WKS and will be further described in the ‘Signs/Symptoms’ section below.
However, as research has developed, and the mapping of the brain and its functions has developed researchers are becoming able to pinpoint the structure’s role within memory, such as the amygdala facilitating the encoding of memories to a more significant degree when the event is emotionally arousing (Josselyn, 2010). This means that when an individual suddenly fails to respond to stimuli that normally would have induced a fear response; a neurologist can narrow down their investigation and begin imaging of the amygdala with the expectation of some sort of abnormality (e.g. a lesion or tumour) affecting it’s functioning.
Once more, if further understanding of Memory and some of the key structures involved (such as the ones mentioned above) is required the below link may be of some use.
https://courses.lumenlearning.com/wsu-sandbox/chapter/parts-of-the-brain-involved-with-memory/